
1994/1995 COMMITTEE MEMBERS
Mr Greg Bryson Dr Margaret Cooley Ms Susan Francis Mr Stephen Hunter
Mr Lyndsay Peters Dr Henry Preston Mr Joseph Webster
1996 COMMITTEE MEMBERS
Ms Susan Francis Mr Peter Hobson Mr John Zaunders
5. LYMPHOCYTE IMMUNOPHENOTYPING
CONTENTS
5.1 Introduction
5.2 Specimen Collection, Transport and Integrity
5.3 Specimen Processing
5.4 Controls
5.5 Sample Analysis
5.6 Data Reporting
5.7 Quality Assurance
5.8 References
5.9 Further Reading
5.10 Appendix 1: Gating Control
5.11 Appendix 2: Reference Range Determination
5.1 INTRODUCTION
The aim of lymphocyte immunophenotyping is to enumerate and identify specific sets or subsets of lymphocytes. This phenotypic analysis is usually performed on blood specimens, however other body fluids may have to be examined.
5.2 SPECIMEN COLLECTION, TRANSPORT AND INTEGRITY
5.2.1 Specimen Collection
5.2.1.1 Universal precautions should be strictly observed when collecting blood samples (see 1.1 Safety Guidelines).
5.2.1.2 A total white cell count and differential and/or stained blood film should be performed at the laboratory initiating the request within the time frame specified by the manufacturer of the haematology instrument used. For distant laboratories and dispatch centres, a total white cell count and unstained blood film should accompany each specimen.
5.2.1.3 EDTA anticoagulated blood and bone marrow specimens are suitable if the specimen will be processed within 12 hours of collection.
5.2.1.4 Heparin or ACD anticoagulated blood and bone marrow specimens may be processed within 48 hours of collection.
5.2.1.5 Any specimen over 48 hours old, or unlabelled, or incorrectly labelled or of insufficient volume should be recollected.
5.2.2 Specimen Transport
5.2.2.1 Packaging, labelling and transport of specimens should comply with all current local, state, national and international regulations for the regions through which the specimens will pass.
5.2.2.2 Specimens should be maintained at 18o 22o C in a light proof container.
5.2.2.3 Temperatures below 4oC, and above 30oC must be avoided.
5.2.3 Specimen Integrity
5.2.3.1 Visually inspect the specimen for clots, haemolysis or container defects. Where appropriate, recollect the sample if the specimen shows any visual signs of deterioration.
5.2.3.2 Specimens that are collected or transported outside of these guidelines should be treated with caution. The deficiencies in the sample should be noted and the report should reflect the effect that these deficiencies may have on the results.
5.3 SPECIMEN PROCESSING
5.3.1 The whole blood lysis method is generally recommended because it does not employ density gradients or lengthy centrifugation which may lead to differential losses of specific subpopulations. However, using this procedure assumes that all leucocyte subsets are equally tolerant to the lysis method used.
5.3.2 Several lysing techniques are available. These include water, tris buffered ammonium chloride and hypotonic buffer1,2. Several proprietary lysing reagents are also available from instrument and monoclonal antibody manufacturers. When using commercial reagents, the manufacturer's recommended protocol should always be followed unless data are available confirming that any modifications do not adversely affect results.
5.3.3 Where possible a full blood count and differential must be performed before processing, and the cell concentration adjusted accordingly. One should aim for a cell concentration of 1 x 106/test tube.
Specimens with pronounced leucopaenia may have insufficient cells for flow cytometric analysis, thus requiring a larger volume of sample or a buffy coat preparation. Conversely, normal concentrations of antibody reagents may be insufficient to saturate all binding sites in specimens with a leucocytosis, leading to possible false negative results. Therefore, samples with a leucocytosis may need to be diluted before testing. A balance between cell and antibody concentration should therefore be found.
5.3.4 Any panel of antibodies must include:
5.3.4.1 Gating controls to allow for correction due to contaminating cells and/or particles (see 5.10 Appendix 1: Lymphocyte Gating).
5.3.4.2. Isotype controls appropriate for the antibodies in the panel.
5.3.4.3. A suitable panel of antibodies for investigating the presumptive diagnosis. The selection of antibodies used in the panel should be referenced.
5.3.5 Any deviation from the manufacturer's recommended protocol should be documented in a laboratory protocol book and only used when mean channel fluorescence or aberrant cell populations are not being studied. Such deviations should show that the results are comparable with those obtained using the recommended procedure.
NOTE: Excess reagent may cause increased nonspecific staining of negatives and may result in decrease of positive/negative resolution.
5.4 CONTROLS
5.4.1 Isotype controls are recognised as an essential part of any monoclonal antibody panel for the purpose of establishing levels of non specific binding and autofluorescence.
In many cases the isotype control may not be optimal for controlling non specific fluorescence because of differences in fluorochrome/protein ratio and antibody concentration between the isotype control and the test reagents. This is particularly important in certain causes of leukaemia (in particular, myelomonocytic) where there is a high degree of species crossreactivity due to the presence of Fc receptors. At this time there is no solution to this problem.
5.4.2 A method control must be prepared and run on a daily basis in parallel with patient samples. At a minimum, a positive reagent control should be prepared and run whenever a new batch of any reagent, used in cell preparation and staining is initiated.
5.5 SAMPLE ANALYSIS
5.5.1 Sample order. All control specimens should be run first and checked, before running the patient samples according to laboratory priority.
5.5.2 Test order within any panel. The first tube should be a gating control to maximise the cells of interest and minimise contamination. The appropriate isotype controls should be run next and then followed by the subsequent test panel to investigate the provisional diagnosis.
5.5.3 Assessment of specimen viability is desirable however because of biohazard concerns, it is recommended that all samples be appropriately fixed prior to analysis on the flow cytometer. It is not presently possible, on a routine large scale basis, to distinguish those cells which were non viable prior to fixation. However, this can be performed using ethidium monoazide (EMA)1.
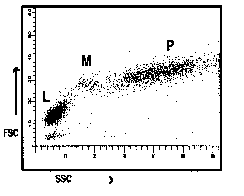
5.5.4 The definition of a lymphocyte gate is shown in Figures 1a and 1b.
Figure 1a Figure 1b
Figures 1a and 1b Representation of common ways of displaying correlated low angle versus 90o angle light scatter seen from lysed whole blood preparations. (L = predominantly lymphocytes, M = predominantly monocytes, P = predominantly polymorphonuclear leucocytes).
5.5.5 Set leucocyte gates as broadly as possible consistent with acceptable levels of contamination to minimise contaminating cells and maximise the inclusion of the cells of interest (see 5.10 Appendix 1:Lymphocyte Gating).
5.5.6 Each laboratory should establish limits of contaminating cells and debris, based on documentation that their inclusion does not significantly affect the measurement of interest. If levels of contamination exceed established laboratory limits, the corrective actions taken are to adjust the light scatter gates and reanalyse the immunofluorescent correlated two colour plot.
Typical satisfactory values for lymphocytes are 95% (minimum 90%) of all lymphocytes and 90% (minimum 85%) purity in the gate as determined by CD45 FITC/CD14 PE gating control (see 5.10 Appendix 1: Lymphocyte Gating).
5.5.7 If levels of contamination by non lymphocytes cannot be minimised to within acceptable limits, then test results may be suspect.
If this contamination cannot be explained by reinterpretation of the data or by clinical diagnostic reasons then a second specimen should be requested.
5.5.8 Count at least 2000 gated events in each sample. This number assures with 95% confidence that the result is within 2% of the "true" value (binomial sampling). NB: This sample mode assumes that the variability of determining replicates is < 2%.
5.5.9 The counting of 2000 gated events to ensure reasonable statistical confidence may not be achievable in severely leucocytopaenic specimens.
5.5.10 Most commercially available directly conjugated reagents give good resolution between low intensity negative and higher intensity positive cell populations. When simultaneous two colour immunofluorescent correlated data is analysed boundaries must be set to define four distinct regions: cells labelled with neither antibody, cells labelled with antibody #1 but not antibody #2, cells labelled with antibody #2 but not #1 and cells labelled with both antibodies.
5.6 DATA REPORTING
5.6.1 Report all unique patient identifiers including name/code, medical record number, laboratory ID/accession number and collection date/time as well as print date/time.
5.6.2 Report all data in terms of cluster of differentiation (CD) with a short description of the main antigen recognition characteristics3.
5.6.3 For unclustered antibodies, report the clone name with a short description of the main antigen binding characteristics.
5.6.4 For blood specimens report all data as a percentage and absolute number of the population of interest within the gate as determined by the gating control.
5.6.5 Report data from all relevant antibody phenotyping combinations with corresponding reference limits of expected normal values, e.g. CD3+8+ Suppressor/Cytotoxic T Cells ± % and/or ± absolute values.
5.6.6 Each laboratory should establish reference limits for the antigens being tested (see 5.11 Appendix 2: Reference Range Determination).
5.7 QUALITY ASSURANCE
5.7.1 Analysis should include internal reliability checks of results, including:
5.7.1.1 Optimally, the sum of CD3+% plus CD19+% plus CD3-CD16+ and/or CD56+ (the " lymphosum"4) should equal the purity of lymphocytes in the gate ± 5%, with a maximum variability of £ 10%. If the data are corrected for lymphocyte purity, then the lymphosum should be between 95 and 105% (minimally 90-110%).
5.7.1.2 Optimally, the sum of the CD3+CD4+% plus CD3+CD8+% should be no more than 5% more than the CD3+%, and no more than 10-15% less than the CD3+%, depending on the number of g/d-TCR+CD3+ cells present.
5.7.1.3 Replicate results within a panel (e.g. CD3+%) for the same sample should be within 5% of each other for FS v SS gating or within 3% for CD45 v SS gating.
5.7.1.4 Light scatter patterns should be examined for each tube within the panel for variation from tube to tube. Similarly, the number of gated events and/or time to collect data should not vary greatly from tube to tube.
Potential sources of error which are not necessarily covered by the above reliability checks may include inappropriate gating leading to exclusion of relevant cells, tubes in a panel run in the wrong order, inappropriate cutoffs between negative and positive cells and calculation or transcription errors. Individual laboratories may require procedures to cover such possibilities.
5.7.2 Each laboratory should determine the level of test variability by preparing and analysing at least six replicates. This will provide a basis when changes to methodology are introduced.
Example 1: A sample control measure is the lymphosum (see 5.7.1.1).
Example 2: Tube to tube variation can be monitored by the inclusion of the same antibody in separate tubes within the one patient test series.
5.7.3 Regulatory bodies currently require that a laboratory keeps all equipment maintenance and calibration records, staff training records, up-to-date method protocols, daily operator/reagent records, verification of transcription of results from machine printouts, procedures for amendment of results and checks by supervisors/pathologists.
5.7.4 Where possible, the laboratory should belong to and participate in a recognised external quality assurance program with regular review of the results.
5.8 REFERENCES
1. Muirhead KA, Wallace PK, Schmitt TC, Rescatore RL, Ranco JA, Horan PK. Methodological considerations for implementation of lymphocyte subset analysis in a clinical reference laboratory. Ann NY Acad Sci 1986;468:113127.
2. Loken MR, Meiners H, Terstappen LWM. Comparison of sample preparation techniques for flow cytometric analysis of immunofluorescence. Cytometry Supplement 1988;2:53.
3. Schlossman SF, et al. Leucocyte typing V. White cell differentiation antigens. Oxford University Press, 1995.
4. Landay A, Auer R, Duque R et al. Quality assurance and immunophenotyping of peripheral blood lymphocytes; Tentative guideline. National Committee for Clinical Laboratory Standards 1992;Guideline No H42T.
5. Nicholson, JKA, Hubbard, M and Jones, BM. Use of CD45 Fluorescence and side-scatter characteristics for gating lymphocytes when using the whole blood lysis procedure and flow cytometry. Cytometry 1996;26:16-21.
6. Winke P, Statlan BE. Reference values. In:Henry JB ed. Clinical diagnosis and management by laboratory methods. Philadelphia: WB Saunders, 1979:2952.
7. Martin HF, Gudzinowicz BJ, Fanger H. Normal values in clinical chemistry. New York: Marcel Dekker, 1975:102236.
8. Henry RJ, Cannon DC, Winkelman JW. Clinical chemistry. Principles and technics. New York: Harper and Row, 1974:343371.
9. Edward BS, Altobelli KK, Nolla HA, et al. A comprehensive quality assessment approach for flow cytometric immunophenotyping of human lymphocytes. Cytometry 1989;10:443441.
10. McCarthy RC, Fetterhoff TJ. Issues in quality assurance in clinical flow cytometry. Arch Pathol Lab Med 1989;113: 658666.
5.9 FURTHER READING
1. CDC. Revised guidelines for the performance of CD4+ Tcell determinations in persons with human immunodeficiency virus (HIV) infection. MMWR 1994;43:RR-3.
5.10 APPENDIX 1: GATING CONTROL
An example of a gating control for lymphocytes where contamination cell types include but are not limited to the following:
1. Nonleukocytes (e.g. CD45 negative cells; potential false negatives; see Figures 2a and 2b)
2. Monocytes (e.g. CD14 positive cells; potential false negatives or positives; see Figures 2a and 2b)
3. Monocytes + granulocytes (e.g. CD13 positive cells; potential false negatives or positives)


Figure 2a Ungated on lymphocytes by scatter Figure 2b Gated on lymphocytes by scatter
Figures 2a and 2b Lymphocyte gating utilising CD14 and CD45 (1 = nonleukocytes, 2 = polymorphonuclear leukocytes, 3 = lymphocytes, 4 = monocytes). Lymphocyte gates should be adjusted to maximise number of cells in region 3 and minimise cells in regions 1, 2, and 4.
CD45 fluorescence v side scatter gating and CD3 fluorescence v side scatter gating have been validated for the measurement of CD4+ and CD8+ subsets of T lymphocytes5, but validation for other subsets is presently unknown.
5.11 APPENDIX 2: DETERMINATION OF REFERENCE RANGES
5.11.1 Definitions
Reference values: Set of values for any measured quantity.
Reference interval: Classically, the range of values found in 95% of a reference population of healthy individuals without overt clinical disease.
NOTE: Age, sex and race are factors known to influence reference intervals.
5.11.2 Procedure for Determining Reference Ranges
Statistical methods, both parametric and nonparametric, and graphical methods are discussed in detail in references 6-8. Only a brief summary of the steps involved is presented here.
5.11.2.1 Parametric methods
The steps of parametric methods are to:
1. Collect data on randomly chosen set of representative individuals (e.g. 50 healthy individuals).
2. Inspect frequency distribution of values obtained.
3. If frequency distribution is Gaussian, use appropriate statistical techniques to estimate 95% confidence interval and use endpoints of interval as the reference range.
4. If frequency distribution is nongaussain, back transform endpoints of 95% confidence interval to obtain reference range, (e.g. log X, of (X + C), square root X, arcsin X) and proceed as in step 3.
5. If no satisfactory transformation can be identified, use nonparametric methods which do not depend on the exact distribution of the data.
5.11.2.2 Nonparametric methods
1. Collection of data on randomly chosen set of representative individuals.
2. Arrangement of data in ascending or descending order.
3. Use of appropriate nonparametric techniques to identify desired limiting percentiles (e.g. 2.5 and 97.5) to desired confidence level.
Nonparametric methods are most appropriate when data does not show a Gaussian distribution and cannot be so transformed. However, they are very sensitive to outliers, and final ranges chosen may be highly dependent on methods used for removing outliers68.
5.11.3 Pitfalls in Determining Flow Cytometric Reference Ranges
Each laboratory should determine its own reference range using its particular preparation method and instrumentation because significant laboratory to laboratory differences related to these variables have been reported.
However, quite large data sets are technically required to carry the above described methods for reference range determination, typically >300 for parametric methods and >120 for establishing a nonparametric interval with 90% confidence. Until more standardised methodology allows pooling of data among laboratories (hence this document), this is clearly an unrealistic expectation.
Other confounding variables besides sample size have been described9,10.
One practical solution to the dilemma is to accumulate and analyse reference data in smaller sets (e.g. 1020 individuals), which can then also be pooled and analysed. If the last two sets of pooled data are found to give the same reference range within experimental error, this gives increased confidence that the reference range selected is not unduly affected by the small sample size.