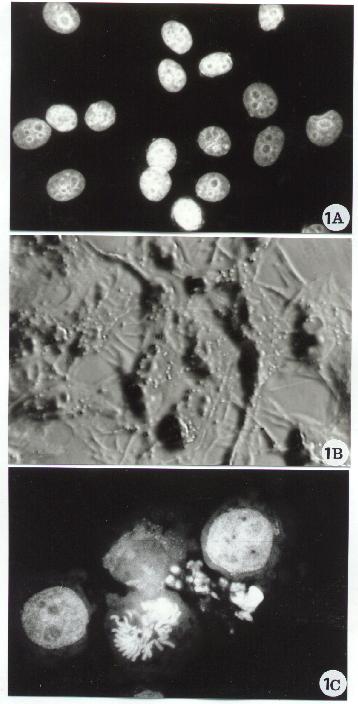
Fig. 1
Subcellular effects induced by physical, chemical and biological agents are often similar and provide some clue to the mechanisms underlying the structural modifications leading to cell death. These changes can also be experimentally induced in vitro by using specific "injury-tools" and can be evaluated by immunochemical and ultrastructural analyses. These models, although semplified with respect to the in vivo situations, can provide useful information for the understanding of the apoptotic process. However, several questions remain unanswered regarding i) the possible relationships between cell structure and function; ii) the possibility of detecting target structures or organelles related to precise pro-apoptotic stimuli; iii) structural differences and homologies between various injury models in various cell histotypes; iv) the ability of a cell to recover (from a precise morphological marker of injury come back to a normal morphology) and v) the point of "no return" and the consequent identification of the boundary line between cell injury and cell death.
In this brief review, the main morphological approaches will be described and some aspects of apoptotic cell death will be shown. The figures are proposed as a sequence of pictures to consult, to look at for comparison or as simple reference material: a sort of small atlas.
The techniques herein described are listed as follows:
B) In situ immunocytochemistry
A) Light microscopy
A1. Introduction
a) Cells Growing In Suspension
Aa.2.1 Materials
2. Slides (25.4 x 76.2 mm)
3. PBS
4. 3.7% paraformaldehyde in PBS
5. 0.5% Triton X-100 in PBS
6. Hoechst 33258
. Wash the coverslips with distilled water (dH2O) to remove completely the acetone.
. Place the coverslips on a clean paper sheet separing each others. Leave the coverslips dry at air.
. Gently spread 20 ml polylysine solution (0.1 mg/ml dH2O) on the coverslips surface.
. Leave the polylysine drops on the coverslips dry at room temperature.
2. Remove the supernatant and resuspend the pellet in 5 ml 3.7% paraformaldehyde in PBS. Leave the sample at room temperature for 15 min.
3. Centrifuge cells at 1000 rpm for 5 min.
4. Remove the supernatant and resuspend the pellet in 5 ml PBS. Centrifuge sample at 1000 rpm for 5 min.
5. Resuspend the pellet in 20-30 m l PBS (depending on the number of cells) and deposit it on polylysine-coated coverslip. Leave the cells adhere on coverslip for 30 min at room temperature.
6. Wash the coverslip twice with PBS.
7. Gently add 0.5% Triton X-100 and leave for 5 min at room temperature. Rinse three times with PBS.
8. Incubate the coverslip with Hoechst 1 µg/ml for 30 min at 37°C.
9. Rinse the coverslip with PBS and mount on the slide with glycerol-PBS (1:1).
10. Observe the sample by a fluorescence microscope (U.V. light).
b) Cells growing in monolayer: detached cells (i.e., following apoptotic treatments).
Ab.2.1. Materials
2. Slides (25.4 x 76.2 mm)
3. PBS
4. 3.7% paraformaldehyde in PBS
5. 0.1% Triton X-100 in PBS
6. Hoechst 33258
Ab.2.2. Methodology
2. Centrifuge the detached cells at 1500 rpm for 10 min. Wash once with PBS. Centrifuge sample at 1500 rpm for 10 min.
3. Resuspend the pellet in 20 ml PBS and deposit it on polylysine-coated coverslip. Leave the cells adhere on coverslip for 30 min at room temperature.
4. Wash the coverslip twice with PBS.
5. Gently add 0.1% Triton X-100 and leave for 5 min at room temperature. Rinse three times with PBS.
6. Incubate the coverslip with Hoechst 1 1 µg/ml for 30 min at 37°C.
7. Rinse the coverslip with PBS and mount on the slide with glycerol-PBS (1:1).
8. Observe the sample with a fluorescence microscope (U.V. light).
c) Cells growing on coverslips
(or remaining adhering to the substrate).
Ac.2.1. Materials
2. PBS
3. 3.7% paraformaldehyde in PBS
4. 0.5% Triton X-100 in PBS
5. Hoechst 33258
3. Place the coverslips on a clean paper sheet separing each others. Leave the coverslips dry on air.
4. Put the coverslips into a heat-resistant glass bottle.
5. Sterilize the coverslips in autoclave for 15 min at 121°C (heat-resistant glass bottle must remain partially opened during sterilization).
6. After sterilization, open the heat-resistant glass bottle in the sterile cup. Leave the coverslips dry with U.V. ray.
7. Close the heat-resistant glass bottle and leave it in the sterile cup.
2. Directly add to the sample the 3.7% paraformaldehyde in PBS. Leave for 20 min at room temperature.
3. Remove the fixative and rinse three times with PBS.
4. Directly add to the sample O.5% Triton X-100 and leave for 5 min at room temperature. Rinse three times with PBS.
5. Incubate the coverslip with Hoechst 1 µg/ml for 30 min at 37°C.
6. Rinse the sample with PBS and mount on the slide with glycerol-PBS (1:1).
7. Observe the sample with a fluorescence microscope (U.V. light).
Samples can also be stained with propidium iodide (fixing cells with ethanol 70% at +4°C for 30 min) or acridine orange 0.1% (fixing the cells with ethanol) in citrate buffer (pH 3).
B) In situ immunocytochemistry
B1.Introduction
2. Tris buffer solution (TBS) pH 7.6
3. poly-L-lysyn (Sigma)
4. Acetone (100%)
5. Ethanol (80% in DW)
6. Methanol (70% in DW)
7. Tunel mixture (Boehringer Mannheim)
8. Mouse-anti-FITC Ab (DAKO)
9. RAM (DAKO)
10. APAAP (DAKO)
11. DAB+developing solution (DAKO)
12. Fast red+developing solution (DAKO)
13. NBT+developing solution (DAKO)
14. Aquamount (BDH)
15. OCT
B2.2.Methodology
*Cytospin preparation:
Carefully wipe slide around the cell spot and dry at room temperature .
Fix the slide sample in absolute acetone for 10 min at 4°C.
Wash with Tris buffer Solution (TBS)(pH 7.6) twice
Dry and saturate the sample with 10% Rabbit serum or FCS or AB human serum as appropriate.
Cover with Tunel solution and incubate for 60 min at 37°C humid atmosphere.
Wash with TBS twice.
Dry and cover with the mouse anti-FITC Ab and incubate for 60 min at room temperature in dark condition.
Wash twice with TBS.
Dry, cover with linking antibody (usually a Rabbit anti-mouse for both the PAP and APAAP method) and incubate for 30 min at room temperature.
Wash twice with TBS.
Dry, cover with APAAP or PAP as appropriate (for Tunel staining we prefer APAAP allowing the use of a wider panel of chromogens, particularly dark chromogen as NBT or fast blue) and incubate for 45 min at room temperature.
Wash twice with TBS.
Dry, cover specimens with developing solution with the substrate-chromogen standard composition suitable for either PAP (0.05% 3,3-diaminobenzidine tetrahydrochloride color reaction, DAB) or APAAP (Fast blue or BCIP/NBT for the dark blue staining, Fast red for red staining + N,N-dimethyl-formamide) and incubate 10 to 30 min.
Block reaction with TBS for 10 min.
Coverslip with aqueous mounting media (Aquamount).
To obtain the best cell morphology the use of chamber slides is recommended. Of course in this case the amount of Tunel mixture as well as of the other reagents highly exceeded that used for cytospin preparation (roughly 5 folds more for chamber slides). Therefore, we use this technique precisely when we have some problem in identifying some morphological parameters. The method is roughly the same of the cytospin preparation, with the only exception of the number of cells used, the technique to attach non-adherent cells on L-polylysin-covered chamber-slide and the permeabilization procedure (unfortunately acetone cannot be used with plastic materials):
treat each chamber with methanol 70% or ethanol 80% for 10 min at +4°C.
wash and follow the same procedure as in (a).
The technique is roughly the same described in (a) with the only exception of the preliminary procedures for collecting, cutting and treating tissue specimens. In particular:
place at -80°C.
cut 4-5 m sections using a -25°C cryostat.
air-dry for 2h to overnight
wash with PBS1x to remove OCT
dry and fix with 100% cold acetone for 10 min at +4°C
wash and air-dry.
carefully wipe slide around the section
follow the same procedure as in (a).
C1. Introduction
Cell lines which grow in monolayer (or dispersed cells seeded on polylysine-coated coverslips) are fixed with 2.5% glutaraldehyde in 0.1M cacodylate buffer. This fixation procedure can induce some changes on the cell surface. To avoid artifacts fixative has to be gently dropped on the samples. After post-fixation in 1% osmium tetroxide, cells are dehydrated through graded ethanols and critical point dried in CO2. The critical point-drying technique provides a much better means of avoiding the damaging effects of surface tension forces than does air drying. The specimen is thus transferred into a closed pressure vessel where it is first infiltrated and surrounded by liquid CO2. The temperature and pressure within the vessel are then raised above the critical values at which point, the critical point, the liquid and gas phases have the same density. After passing the critical point, the pressure is slowly reduced so that the recondensation of the fluid through adiabatic cooling is avoided.
To allow scanning electron microscopy observation the specimens are
then normally coated with a conducting layer to inhibit charging. Sputter
coating by gold has the advantage of requiring only a low vacuum and has
been proved to be satisfactory for observational work.
a) Cells growing in suspension
Ca.2.1. Materials
3. 2.5% glutaraldehyde in 0.1M cacodylate buffer
4. 0.2M cacodylate buffer (pH 7.4)
5. 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer
6. Graded ethanols
7. Absolute ethanols
8. Stubs
9. Silver print
Fixation
2. Remove the supernatant and resuspend the pellet in 5 ml 0.1M cacodylate buffer. Centrifuge sample at 1000 rpm for 5 min.
3. Remove the supernatant and resuspend the pellet in 5 ml 2.5% glutaraldehyde in 0.1M cacodylate buffer. Leave the cells fix for 20 min at room temperature.
4. Centrifuge cells at 1000 rpm for 5 min.
5. Remove the supernatant and wash the cells twice with 5 ml 0.1M cacodylate buffer.
After fixation with glutaraldehyde, samples can be kept at +4°C in 0.1M cacodylate buffer for several days.
6. Centrifuge cells at 1000 rpm for 5 min.
7. Resuspend the pellet in 20-30 ml 0.1M cacodylate buffer and deposit it on polylysine-coated coverslip. Leave the cells adhere on coverslip for 20 min at room temperature.
8. Delicately add 0.1M cacodylate buffer.
Remove 0.1M cacodylate buffer and add 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer. Leave the cells fix for 30 min at room temperature.
9. Wash the coverslip three times with 0.2M cacodylate buffer. After post-fixation with OsO4, samples can be kept at +4°C in 0.2M cacodylate buffer for 1-2 days.
11. Remove the 30% ethanol. Add 50% ethanol. Leave for 5 min and remove (repeat three times).
12. Remove the 50% ethanol. Add 70% ethanol. Leave for 10 min and remove (repeat twice). At this point the dehydration procedure may be stopped for up to one week. Samples can be kept in 70% ethanol at +4°C.
13. Remove the 70% ethanol. Add 85% ethanol. Leave for 15 min and remove (repeat twice).
14. Remove the 85% ethanol. Add 95% ethanol. Leave for 15 min and remove (repeat twice).
15. Remove the 95% ethanol. Add absolute ethanol. Leave for 10 min. Repeat three times leaving the samples always covered by ethanol during preparation of CPD. Never leave the samples dry.
16. Dehydrate samples by CPD.
17. Glue the coverslips onto stubs with silver print. Let dry about 20 min at room temperature and overnight at 37°C.
18. Cover the samples with gold (thickness 20 nm) using the sputter coater.
b) Cell lines which grow in monolayer
(on the coverslips)
C.b.2.1. Materials
1. coverslips
2. 0.1M cacodylate buffer (pH 7.4)
3. 2.5% glutaraldehyde in 0.1M cacodylate buffer
4. 0.2M cacodylate buffer (pH 7.4)
5. 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer
6. Graded ethanols
7. Stubs
8. Silver print
C.b.2.2. Methodology
2. Directly add to the sample the 2.5% glutaraldehyde in 0.1M cacodylate buffer. Leave the cells fix for 20 min at room temperature.
3. Remove the fixative and rinse three times with 0.1M cacodylate buffer.
After fixation with glutaraldehyde, samples can be kept at +4°C in 0.1M cacodylate buffer for several days.
4. Remove 0.1M cacodylate buffer and add 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer. Leave the cells fix for 30 min at room temperature.
D) Transmission Electron Microscopy (TEM)
D1. Introduction
Ultrathin sections. Cell monolayers (and cells recovered from the surnatant and gently centrifuged) can be fixed in glutaraldehyde and post fixed in OsO4. Different protocols can be used depending on the type of sample considered. In particular, cell pellets or monolayer are easily fixed (only vapours are sometime sufficient) while tissues need longer times of exposure to fixative. However, glutaraldehyde is capable of penetrating very poorly inside tissues (no more then 2mm).
Specific beems can be used to collect both these samples. In particular, cell monolayers are embedded directly in the culture flasks after at last three different embedding steps. A first step is performed by using low resin concentrations. Other two or three steps are then performed by using increasing concentrations of embedding resin. Finally, absolute resin is used in two embedding steps of one-two hours.
D2.1. Materials
2. 2.5% glutaraldehyde in 0.1M cacodylate buffer
3. 0.2M cacodylate buffer (pH 7.4)
4. 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer
5. Agar 2% in dH20
6. Graded ethanols
7. Absolute ethanol
8. Propylene oxide
9. Agar 100 resin
D2.2. Methodology
Fixation:
*Dispersed cells
2. Remove the supernatant and resuspend the pellet in 5 ml 0.1M cacodylate buffer. Centrifuge sample at 1000 rpm for 5 min.
3. Remove the supernatant and resuspend the pellet in 5 ml 2.5% glutaraldehyde in 0.1M cacodylate buffer. Leave the cells fix for 20 min at room temperature.
4. Centrifuge cells at 1000 rpm for 5 min.
5. Remove the supernatant and wash the cells twice with 5 ml 0.1M cacodylate buffer.
After fixation with glutaraldehyde, samples can be kept at +4°C in 0.1M cacodylate buffer for several days.
6. Centrifuge cells at 1000 rpm for 5 min.
7. Remove the supernatant and resuspend the pellet in 3-4 ml 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer. Leave the cells fix for 1h at room temperature.
8. Centrifuge cells at 1000 rpm for 5 min.
9. Remove the supernatant and wash the cells twice with 5 ml 0.2M cacodylate buffer.
After post-fixation with OsO4, samples can be kept at +4°C in 0.2M cacodylate buffer for 1-2 days.
10. Centrifuge cells at 1000 rpm for 5 min.
11. Remove the supernatant and embed the pellet in 2% agar.
12. Cut the embedded pellet in pieces of about 1 mm3. Put the pieces in a small glass bottle.
*Cell lines growing in monolayer
1. Remove the medium and wash the cells twice with 0.1M cacodylate buffer.
2. Directly add to the sample the 2.5% glutaraldehyde in 0.1M cacodylate buffer. Leave the cells fix for 20 min at room temperature.
3. Remove the fixative and rinse twice with 0.1M cacodylate buffer. After fixation with glutaraldehyde ,samples can be kept at +4°C in 0.1M cacodylate buffer for several days.
4. Remove 0.1M cacodylate buffer and add 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer. Leave the cells fix for 1h at room temperature.
5. Remove the fixative and add 0.2M cacodylate buffer. Leave for 5 min at room temperature and remove (repeat three times). After post-fixation with OsO4, samples can be kept at +4°C in 0.2M cacodylate buffer for 1-2 days.
*Animal tissues
1. Cut the tissue in pieces of about 1 mm3, leaving the samples always covered by 0.1M cacodylate buffer. Never leave the samples dry.
2. Put the tissue pieces in a small glass bottle always covered by 0.1M cacodylate buffer.
3. Remove the buffer and directly add 2.5% glutaraldehyde in 0.1M cacodylate buffer completely covering the tissue pieces. Leave the cells fix for 2h at +4°C.
4. Remove the fixative and add 0.1M cacodylate buffer. Leave for 30 min at +4°C and remove (repeat twice).
5. Remove the buffer and directly add 1% osmium tetroxide (OsO4) in 0.2M cacodylate buffer completely covering the tissue pieces. Leave the cells post-fix for 30 1h at +4°C.
6. Remove the fixative and add 0.2M cacodylate buffer. Leave for 5-10 min at room temperature and remove (repeat three times). After post-fixation with OsO4, samples can be kept at +4°C in 0.2M cacodylate buffer for 1-2 days.
Dehydrate the samples through graded ethanols as follows:
| Cultures | Monolayers | Tissue | |
| 30% ethanol | 2 x 10 min | 2 x 5 min | 2 x 10 min |
| 50% ethanol | 2 x 10 min | 2 x 5 min | 2 x 10 min |
| 70% ethanol | 2 x 10 min | 2 x 5 min | 2 x 10 min |
| 70% ethanol | 2 x 10 min | 2 x 5 min | 2 x 10 min |
At this point the dehydration procedure may be stopped. Samples can be kept in 70% ethanol at +4°C.
| 85% ethanol | 1 x 10 min | 1 x 10 min | 1 x 10 min |
| 95% ethanol | 1 x 30 min | 1 x 10 min | 1 x 30 min |
| Absolute ethanol | 3 x 20 min | 3 x 10 min | 2 x 30 min |
| Propylene oxide
(or toluene) |
1 x 10 min | 1 x 20-30 min |
Inclusion
Include the samples as follows:
| dispersed cells | tissues | |
| Propylene oxide/Resin 2:1 | 1 x 1h | 1 x 1h |
| Propylene oxide/Resin 1:1 | 1 x 1h | 1 x 1h |
| Propylene oxide/Resin 1:2 | 1 x 2h | 1 x 2h |
| Absolute resin | 2 x 1h | 1 x 2h |
| Absolute resin into the beems | 1 x 1h | 1 x 2h |
| Monolayer | |
| Absolute ethanol/Resin 2:1 | 1 x 1h |
| Absolute ethanol/Resin 1:1 | 1 x 1h |
| Absolute ethanol/Resin 1:2 | 1 x 1h |
At this point the inclusion procedure may be stopped for an overnight. Samples can be kept in absolute ethanol/Resin 1:2 at +4°C.
| Absolute resin | 2 x 1h |
| Absolute resin with the beems | 1 x 1h |
Monolayers: fill beems with absolute resin and put them upside down
on the cell layer.
Polymerization
Monolayers: gently detach the beems. They will detach with small fragments of polymerized resin. A dark surface will indicate the presence of cells.
Polymerize at 60°C for 48h.
3. Commentary
3.1. Background information
The cytochemical and immunocytochemical techniques allow to investigate morpho-functional correlates of the various apoptotic pathways. Double staining procedures can be often used to correlate both apoptotic cells to their phenotype, cytokine production, virus and microbial infections, cytoskeletal organization, transcription factor expression, calcium ions, redox alterations and so on. Moreover, in situ immunocytochemical techniques are useful to investigate the distribution of the various cell proteins (e.g. adhesion molecules) both at the single cell level and at the cell-to-cell contact sites. In other words, the in situ evaluation of apoptosis through immucyto- and histochemistry provides unique information for studies aimed to identify apoptotic mechanisms. Electron microscopy, TEM in particular, should be avoided for routinary studies.
2. Often the morphological features of apoptosis can be confused or overlap those typical of necrosis. For instance: the so-called secondary necrosis is difficult to be recognized and, in any case, necrosis should also be always evaluated; superaggregation of chromatin could often occur in situations other than apoptosis; lymphocytes very rarely undergo chromatin clumping although undergoing apoptosis; surface blebbing as well as surface smoothing are common markers of necrosis as well as of apoptosis, etc. Moreover, because apoptosis is an asyncronous process, all the apoptotic stages are usually detectable, earlier (light chromatin marginalization) and latest stages (residual bodies).
3. In general, all the in situ methods need years of experience for evaluating data. Some problems may come in quantifying positive cells both in cytospin or polylYsine preparations and in cryostat sections, and some counterstaining procedures and/or double staining with cell specific markers may help in better distinguish cells in the various stages of apoptosis. In particular, double staining procedures aimed to discriminate apoptotic to proliferating cells are recommended when an effect on cell differentiation, of a given drug or cytokine, may be in progress.
4. The amount of cells represents a critical factor for studies on apoptosis. For dispersed cell analyses (e.g. lymphocytes) few cells are necessary for light microscopy and SEM studies, about 200.000 cells. For TEM analyses, the pellet should be of at least 5-6x106 cells.
5. Advantages in using light microscopy analysis are represented by the time necessary for performing the assay. On the other hand, the pletora of information provided by SEM and TEM approaches, e.g. the study of organelles and cytoskeleton, is wide, may be useful for subsequent studies, e.g. biochemical or molecular, and furnishes important controls for any experimental tasks.
3.4. Time considerations
2. Allen, T.D. Ultrastructural aspects of cell death. In: Perspectives on mammalian cell death. (C.S. Potten, ed.) Oxford University Press, Oxford, pp.39-65, 1987
3. Bellomo G., F. Mirabelli, M Vairetti, F. Iosi, and W. Malorni 1994. Morphological and biochemical investigations on plasma membrane blebbing during cell injury. In: In vitro toxicity indicators, Methods in toxicology, part B: (C.A. Tyson and J.M. Frazier eds.), vol 1, pp. 58-71.
4. Malorni, W., Rivabene, R., Maria Teresa Santini, M.T. and Gianfranco Donelli, G. 1993. N-acetylcysteine inhibits apoptosis and decreases viral particles in HIV-chronically infected U937 cells. FEBS Letters, 327, 75-78.
5. Allen, R.T. 1997 Morphological and biochemical characterization and analysis of apoptosis J. Pharmacol. Toxicol. Methods 37, 215-228, 1997
6. Gold, R., Schmied, M., Rothe, G., Zischlier, H., Breitschopf H., Wekerle, H., Lassmann, H. 1993. Detection of DNA fragmentation in apoptosis: application of in situ nick translation to cell culture systems and tissue sections. J. Histochem. Cytochem. 41: 1023-1030.
7. Negoescu, A., Lorimier, P., Labat-Moleur, F., Drouet, C., Robert, C., Guillermet, C., Brambilla, C. and Brambilla, E. 1996. In situ apoptotic cell labelling by the Tunel method: improvement and evaluation on cell preparations. J. Histochem. Cytochem. 44: 959-968.
8. Fais, S., Burgio, V.L., Silvestri, M., Capobianchi, M.R., Pacchiarotti, A., Pallone, F. 1994. Multinucleated Giant Cells generation induced by IFN-gamma. Changes in the expression and distribution of the intercellular adhesion molecule-1 during macrophage fusion and MGC formation. Lab. Invest: 15: 1033-36.
9. Fais, S., Capobianchi, M.R., Abbate, I., Castilletti, C., Gentile,
M., Cordiali Fei, P., Ameglio, F., Dianzani, F. 1995. Unidirectional budding
of HIV-1 at the site of cell-to-cell contact is associated with co-polarization
of intercellular adhesion molecule-1 and HIV-1 viral matrix protein. AIDS
9: 329-35.
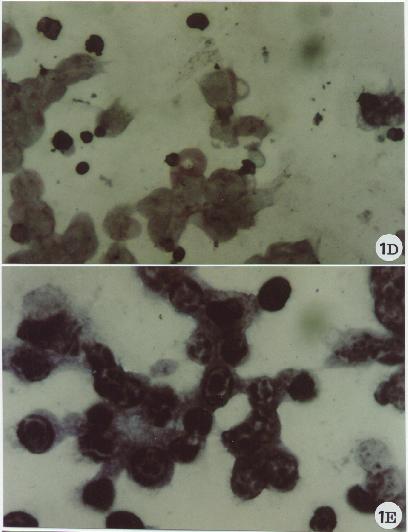
Fig. 1

Fig. 2
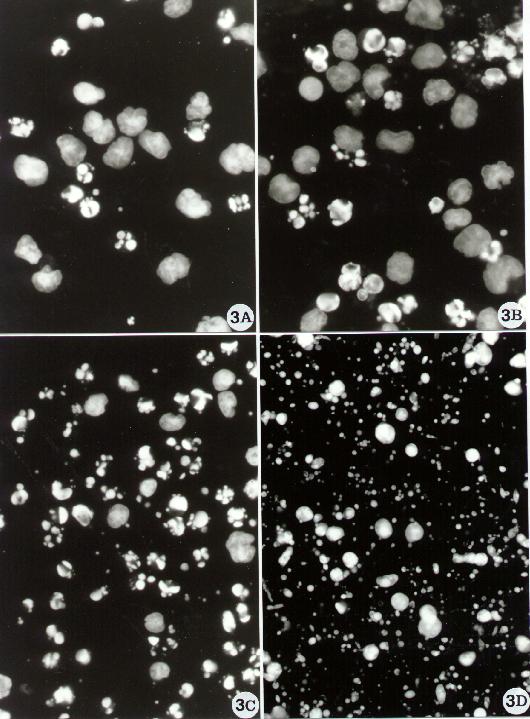
Fig. 3


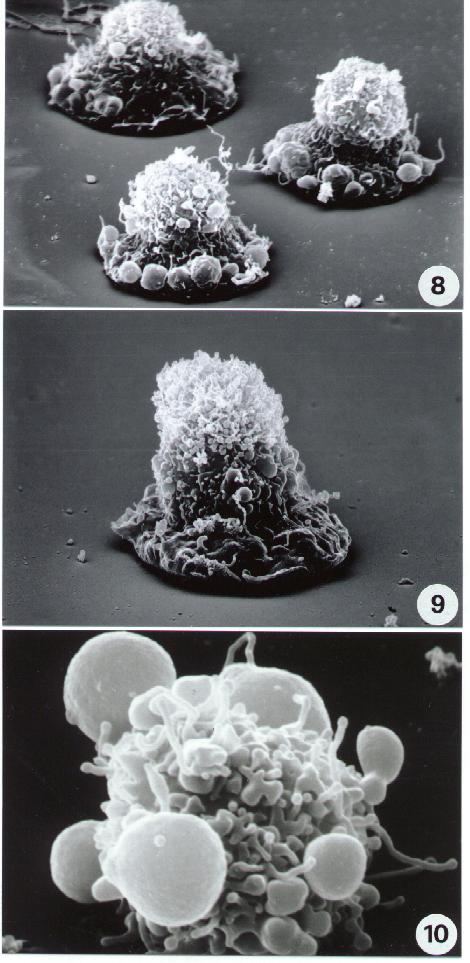
Fig. 5-14
11-14. Scanning Electron microscopy. Different aspects of necrotic cell death. Compare necrotic surface alterations including surface blebbing in (14) with apoptotic ultrastructure (8-10).
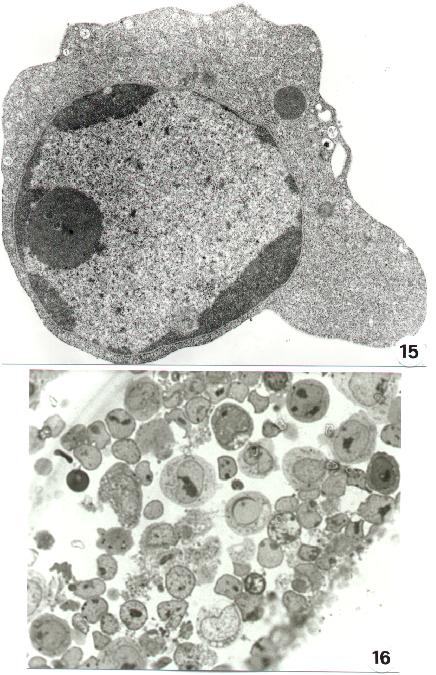
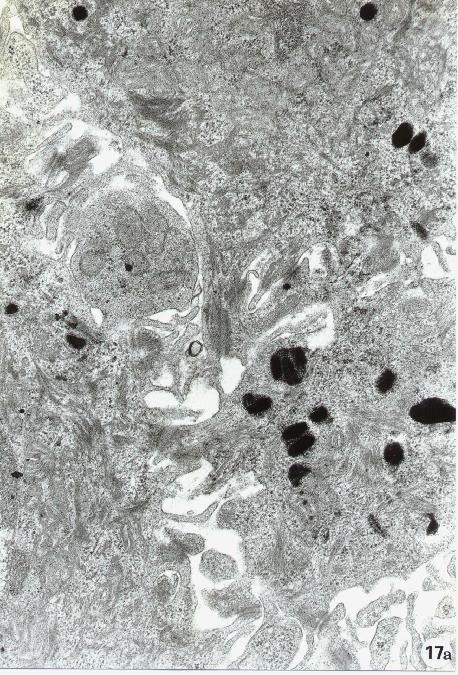
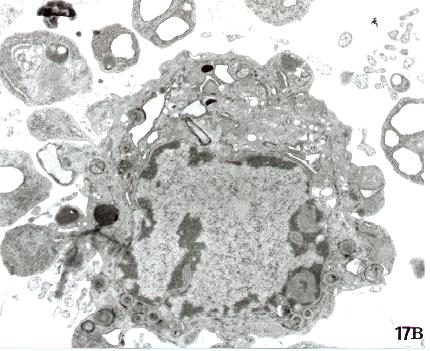
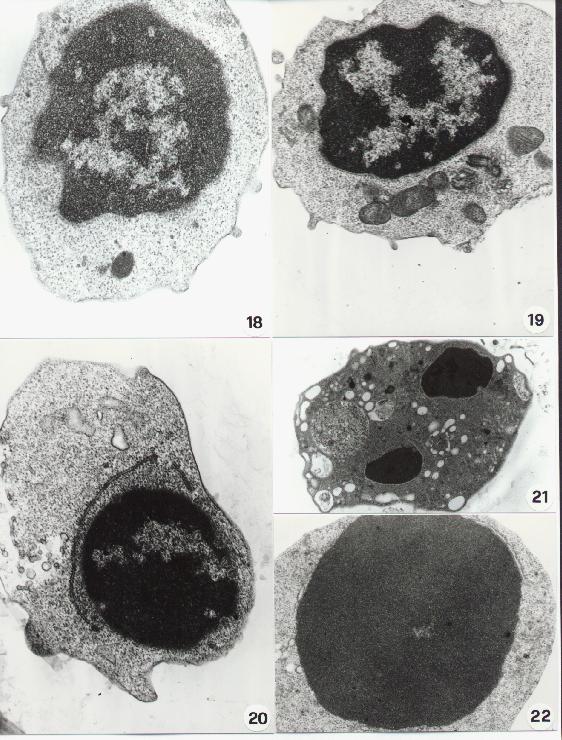
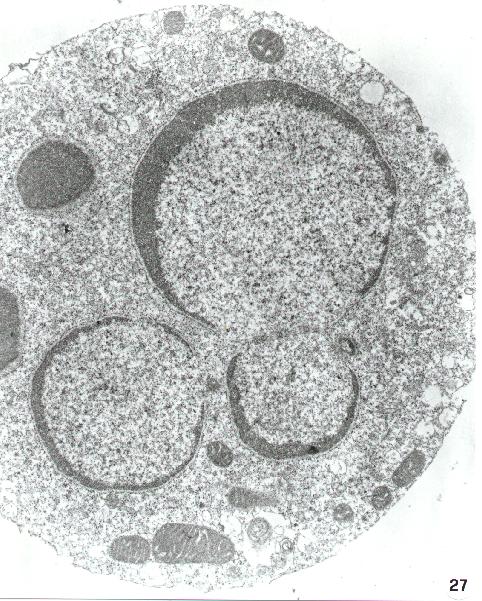
Fig. 15-22
17. Transmission Electron Microscopy. "In vivo" blebbing. Section from a human tissue. Note a surface bleb between two epithelial cells (a). In (b) is shown a surface blebbing in an apoptotic cell.
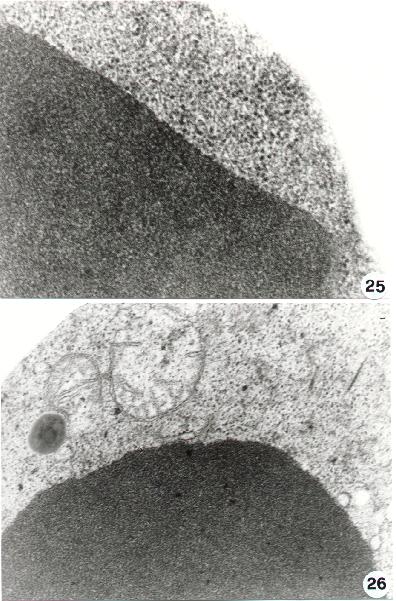
Chromatin aggregation and condensation shows sometimes a paracristalline structure (25). The chromatin "superaggregation" of an apoptotic cells is shown in figure 26. To note the mitochondrial lesions and the lack of nuclear envelop which can be detected at this stage.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}