The apoptotic process is characterized by activation of nucleases which
produce a large number of DNA strand breaks. These breaks can be labelled
by attaching to them biotin- or digoxygenin conjugated dUTP in a reaction
catalysed by exogenous terminal deoxynucleotidyl transferase (TdT-assay)
or DNA polymerase.
Fluorochrome-conjugated avidin or digoxygenin antibodies are used as
second step to label the DNA breaks and more recently, fluorochrome-conjugated
deoxy-nucleotides have been made available for a single-step labelling
procedure. Detection of breaks requires a pre-fixation with a cross-linking
agent, such as paraformaldheyde that unlike ethanol prevents the extraction
of fragmented DNA. Cells are then post-fixed in cold ethanol, labelled
with nucleotides in a TdT catalysed reaction, resuspended in PBS and counterstained
with PI for a simultaneous analysis of cell-cycle and apoptosis.
Materials
Paraformaldehyde 1% in PBS pH 7.4
Ethanol
Potassium Cacodylate
PBS
TRIS-HCl
Triton X-100
BSA
Cobalt Chloride
Terminal Transferase (TdT from Sigma or Boheringer)
FITC-dUTP (or BODIPY-dUTP or CY2-dUTP from Boheringer or Molecular Probes
or Amersham)
RNAse A
Propidium Iodide (PI) stock solution 50 mg/ml in PBS
Equipment
Centrifuge
Incubator
Flow Cytometer
(A1) TdT Staining Solution
10 ml of the reaction buffer (1 M Potassium
Cacodylate pH 7, 125 mM TRIS-Hcl pH 6.6, 1.25 mg/ml BSA)
5 ml of Cobalt Chloride solution (CoCl2
25 mM)
12.5 units of TdT in its storage buffer (usually 0.5 ml)
0.25 nanomoles of FITC-dUTP (or BODIPY-dUTP) in its storage buffer
Add distilled water to 50 ml
(A2) Rinsing Buffer
PBS containing 0.1% Triton X-100 and 5 mg/ml BSA
(A3) PI Staining Solution
Propidium Iodide 5 mg/ml, RNase A 200 mg/ml
in PBS
Methodology:
Wash cells (1x106) in PBS and centrifuge at 200 g for 5 min
Fix cells in 1 ml 1% paraformaldehyde pH 7.4 for 15 min on ice
Centrifuge at 200 g for 5 min and resuspend the cell pellet in 5 ml PBS
Centrifuge and resuspend cells in 0.5 ml PBS
Post-fix the cell suspension in 5 ml ice-cold 70% (vol/vol) ethanol. Leave
cells in ethanol for at least one hour (the cells can be stored in ethanol
at -20°C for several days)
Centrifuge, remove ethanol, resuspend cells in 5 ml PBS and centrifuge
Resuspend the cell pellet in 50 ml of TdT staining
solution (A1)
Incubate cells for 60 min at 37°C
Add 1.5 ml of rinsing buffer resuspend throughly and centrifuge
Repeat rinsing in 1.5 ml of rinsing buffer (A2) and centrifuge
Resuspend cell pellet in 1 ml of PI staining solution (A3)
Incubate for at least 30 min at room temperature in the dark and analyse
cells by flow cytometry
For flow cytometer analysis: construct a dot plot of PI red fluorescence
on x-axis vs. green fluorescence of incorporated nucleotides on y-axis
Set red (PI) fluorescence acquisition in linear mode
Set green (FITC) fluorescence acquisition in logarithmic scale
Set photomultiplier parameters (gain and compensation) appropriately (a
classic example is provided in figure 1)
Run the samples. Collect at least 104 cells/sample
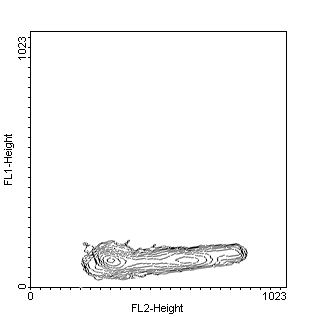
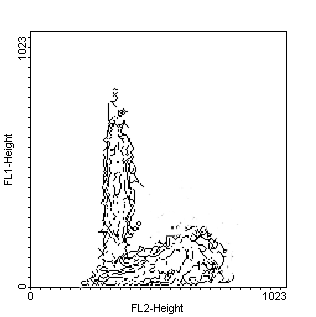
Figure 1.TdT assay in in vitro cultured MCF-7 breast cancer
cells and examined by flow cytometry. Nuclei are counterstained with PI
to analyse the relationships between cell-cycle position and apoptosis.
Contour plot of log-growing cells (left panel) does not show any green
fluorescence. Treatment with genistein (100 mM 12h and subsequent 12 h
release, middle panel) is followed by the appearance of green-stained (apoptotic)
cells. The apoptosis is maximal in the G0-G1 phase of the cell cycle. Continuous
(24 h) exposure to the same agent (right panel) produce apoptosis in both
G0-G1 and G2/M cell-cycle phases.
Alternative methods:
Alternative methods utilize either digoxigenin-conjugated d-UTP followed
by staining with a FITC-conjugated anti-digoxigenin Ab or biotin-conjugated
d-UTP followed by staining with FITC-conjugated streptavidin. These methods
produce a stronger emission in the green (FITC) channels but they increase
the aspecific staining, with a reduced signal/background ratio.
Commentary
Background informations
This is the most specific method for evaluation of early phases of apoptosis
Commercial kits (Boheringer and Amersham) for this assay are available
Since the DNA content of cells is simultaneously measured, the method provides
the unique possibility of analysing the cell-cycle position of apoptotic
cells
An important disadvantage stems from the high cost (particularly when commercially
available kits are used)
Critical parameters
A proper fixation to avoid loss of fragmented DNA (the major source of
staining!!) is essential
Maintain the TdT stock solution at 20°C since its activity is critical
for nucleotide incorporation into DNA strand breaks
Troubleshooting
Necrotic cells and/or cells repairing DNA can present some staining particularly
when indirect methods (see alternative methods) are utilized
Key References
Gavrieli Y, Y Sherman and SA Ben-Sasson. 1992. Identification of programmed
cell death in situ via specific labeling of nuclear DNA fragmentation.
J Cell Biol119:493-501
Gorczyca W, J Gong and Z Darzynkiewicz. 1993. Detection of DNA strand breaks
in individual apoptotic cells by the in situ terminal deoxynucleotidyl
transferase and nick translation assays. Cancer Res53:1945-1951
Darzynkiewicz Z, X Li and J Gong. 1994. Assay of cell viability: Discrimination
of cells dying by apoptosis. In: Flow cytometry (2nd Edition).
Z Darzynkiewicz, JP Robinson and HA Crissman eds. Academic Press, New York,
pp 15-38.