APOPTOSIS vs. NECROSIS
Marco Vitale*, Giorgio Zauli° and Elisabetta Falcieri+
*Dept. Biomedical Sciences and Biotechnologies, University of Brescia, Italy °Institute of Human Anatomy, University of Ferrara, Italy +Institute of Anatomy and Physiology, University of Urbino, Italy.
EMail: vitale@master.cci.unibs.it
It is generally accepted that cell death can either be the consequence
of a passive, degenerative process, or the consequence of an active process.
The former type of cell death is termed necrosis, the latter apoptosis.
Apoptosis was originally identified morphologically. The explosion of studies
on apoptosis in recent years has clarified that it represents the mode
of death that is actively driven by the cell, a complex process that is
indicated as programmed cell death. On the opposite, necrosis represents
a passive consequence of gross injury to the cell. It is morphologically
different from apoptosis, and its physiological consequences are also very
different from those of apoptosis. Being apoptosis a very common phenomenon
during embryogenesis as well as adult life, and a frequent response of
cells to drugs in the course of therapy, its quantitative evaluation represents
an issue of considerable relevance. Flow cytometry is the choice technique
for the quantitation of apoptosis. A multitude of methods have been described
to identify apoptotic cells by flow cytometric analysis (see for review,
Darzynkiewicz Z. et al., Cytometry 27: 1-20, 1997), each of them differently
suitable to different experimental conditions. One general problem that
arises virtually always with the flow cytometric analysis of apoptosis
is the distinction between necrotic and apoptotic cells. Moreover, although
necrotic cells are usually felt as contaminating objects during the quantification
of apoptosis, one might be interested in the quantification of both apoptosis
and necrosis in a given cell population. There is no clear cut parameter
that allows the separation by flow cytometry of necrotic from apoptotic
cells, particularly at their late stages. On the contrary, such a distinction
is immediate by morphological techniques. Even the mere presence of apoptotic
cells in a given population should be always validated by morphological
observations. Therefore, flow cytometry and optical and electron microscopy
all contribute, from different perspectives, to the identification of apoptosis
and to its distinction from necrosis. Therefore, here will be described
some common flow cytometric and morphological procedures for the identification
and measurement of apoptotic cells with a particular emphasis on the discrimination
of apoptotic from necrotic cells.
APOPTOSIS vs. NECROSIS BY FLOW CYTOMETRY
1. Introduction
It may be useful to make some general comments on the flow cytometric
distinction of apoptotic from necrotic cells before describing the methods.
1) A univocal method to distinguish apoptotic from necrotic cells
by flow cytometry does not exist. Rather, it is necessary to discriminate
necrotic cells from apoptotic cells in each of the many flow cytometric
methodologies available to measure apoptosis. Only the scatter method
can apply in combination with most of the other methods to analyze apoptosis
by FACS, which makes this method, that has some limitations per
se, very useful.
2) Given the fact that most methodologies concentrate on the detection
of apoptotic cells, often the problem is to discriminate healthy from necrotic
cells. This can generally be achieved by the scatter method. The combination
of a specific methodology designed to reveal apoptosis and the scatter
analysis of the cell population generally allows a satisfactory distinction
between healthy, necrotic and apoptotic cells.
3) A bigger problem arises with debris. It is of the utmost importance
that everyone using flow cytometry to quantify apoptosis realizes that
the fate of a apoptotic cell in vitro is to give origin to many small apoptotic
bodies, which generally end up as debris at the flow cytometric analysis,
exactly as the final products of cell necrosis. Even if it was possible
to count the apoptotic bodies, this would be useless, since it would be
impossible to know how many apoptotic cells they derive from. Therefore,
all the techniques that we use to enumerate apoptosis, work until the apoptotic
cell is still a cell. Being apoptosis a asynchronous process, it is evident
that we can analyze only a window of the whole process.
4) Morphological analysis is essential to confirm the flow cytometric data.
This is why after the flow cytometric methodologies, a section will follow
in this chapter, in which the distinction between apoptosis and necrosis
will be described by morphological techniques.
2A. First protocol: light scatter
2A.1. Materials
-PBS, pH 7.0
-Flow cytometer with forward (FSC) and side (90°) scatter (SSC)
detection, laser tuned at 488nm wavelength
2A.2. Methodology
1. Put in a tube 1x105 cells in which you want to test the presence of apoptosis, and resuspend them in 300-500 ul of PBS.
2. If possible, put in a tube 2x105 healthy cells of the same type, and resuspend them in 600-1000 ul of PBS. Divide this sample in two, by putting vol/2 in a new tube. Take one of the tubes and freeze and thaw it 3 times, to generate a population of necrotic cells. The intact cells of the other tube will serve as control for healthy cells.
3. If allowed by the type of instrumentation, set the flow cytometer paying particular attention to the light scatter detection system. Use a 488 nm wavelength laser emission. It can be useful to put a 488 nm band pass filter before the SSC photomultiplier (PMT).
4. Analyze the cells by flow cytometry. Start the analysis with the sample that contains healthy cells on the cytogram (FSC vs SSC). Place the population at mid-high FSC values and mid SSC values working on signal gains and PMT voltage. This will allow to have some space on the cytogram at lower FSC values and slightly higher SSC values.
5. Analyze the necrotic cell sample. It is likely that the necrotic population will appear at lower FSC and SSC values, frequently merging with debris.
6. Analyze the sample which should contain apoptotic cells. Depending on the cell type and on the nuclear/cytoplasmic (N/C) ratio of the intact cell, a third population will appear, generally at lower FSC and slightly higher SSC values with respect to healthy cells.
This scheme mainly refers to thymocytes, and does not have a general value, although other cell types may give similar results. Identification of apoptotic and necrotic cells as distinct populations by scatter parameters may not be possible in some cell types, and therefore each cell type must be tested in pilot experiments. It is also advisable to sort (if possible) each population (few hundreds of cells directly on a slide will be sufficient), and look at them under the microscope after Wright or Giemsa staining.
3A. Commentary
3A.1. Background information
When a cell passes trough a laser beam in a flow cytometer, generates light scatter. FSC provides information about cell size, SSC about the cell s morphological complexity. When a cell dies, its morphology changes, and changes in light scatter may reflect these phenomena.
Necrosis: when a cell dies by necrosis, both FSC and SSC tend to increase,
likely as a consequence of cell swelling. However, as a consequence of
plasma membrane damage and leakage of cell constituents, both FSC and SSC
rapidly decrease.
Apoptosis: when a cell dies by apoptosis, its major morphological changes
take place in the nucleus. Only in the late apoptotic stages the cytoplasm
and the plasma membrane are seriously damaged. It has in fact been demonstrated
that the higher is the N/C ratio in a given cell, the better is the distinction
between apoptotic, necrotic and healthy cells by light scatter.
3A.2. Time considerations
The flow cytometric procedure will not take more than 30 min.
3A.3. Key references
1. Zamai, L., Falcieri, F., Zauli, G., Cataldi, A., and Vitale, M. 1993.
Optimal detection of apoptosis by flow cytometry depends on cell morphology.
Cytometry 14: 891.
2. Ornerod, M. G., Cheetham, F. P. M., and Sun, X. M. 1995. Discrimination
of apoptotic thymocytes by forward light scatter. Cytometry, 21: 300.
2B. Second protocol: membrane permeability by propidium iodide
2B.1. Materials
-PBS, pH 7.0
-propidium iodide (PI) 40 ug/ml in PBS (always wear gloves when using PI)
2B.2. Methodology
1. Put in a tube 1x105 cells in which you want to test the presence of apoptosis, and resuspend them in 300-500 ul of PBS.
2. If possible, put in a tube 2x105 healthy cells of the same type, and resuspend them in 600-1000 ul of PBS. Divide this sample in two, by putting vol/2 in a new tube. Take one of the tubes and freeze and thaw it 3 times, to generate a population of necrotic cells. The intact cells of the other tube will serve as control for healthy cells.
3. Centrifuge and resuspend the pellets with the PI solution. Leave the samples at RT for an interval between 30 min and 1.5 hr. This time has to be adjusted on the cell type.
4. Analyze by flow cytometry the healthy cell sample s red fluorescence on a log scale. Place the peak at low fluorescence values (between 100 and 101) working on signal gain and PMT voltage.
5. Analyze the necrotic cell sample. These cells will be all brightly stained by PI, and will appear as a peak at very high fluorescence values.
6. Analyze the sample which may contain healthy, apoptotic and necrotic cells. Apoptotic cells will appear as a dimly fluorescent population.
3B. Commentary
3B.1. Background information
The intact membrane of living cells excludes cationic dyes, such
as PI or trypan blue. Due to their extensive membrane damage, necrotic
cells are quickly stained by short incubations with PI. Apoptotic cells
(with the exception of late apoptoses, which, from this standpoint, behave
as necrotic cells) show an uptake of PI that is much lower than that of
necrotic cells. It is therefore possible to distinguish healthy (PI negative),
apoptotic (PI dim) and necrotic (PI bright) cells from each other. This
method can be usefully combined with the scatter method.
During the method optimization, it is useful to sort (if possible)
a few hundreds cells on a slide and directly look at them under the fluorescence
microscope.
3B.2. Troubleshooting
Usually the distinction between apoptotic and necrotic cells is quite evident, while the discrimination between healthy and apoptotic cells may be difficult. If these two populations tend to merge, increase the time of incubation with PI. If distinction is good but the peak of healthy cells is too high, decrease the time of incubation with PI.
3B.3. Time considerations
The whole procedure will take 3 hr or less, depending on the length of the incubation with PI.
3B.4. Key references
1. Lyons, A. B., Samuel, K., Sanderson, A., and Maddy, A. B. 1992. Simultaneous
analysis of immunophenotype and apoptosis of murine thymocytes by single
laser flow cytometry. Cytometry 13: 809.
2. Vitale, M., Zamai, L., Mazzotti, G., Cataldi, A., and Falcieri, E. 1993.
Differential kinetics of propidium iodide uptake in apoptotic and necrotic
thymocytes. Histochemistry 100: 223.
3. Zamai, L., Falcieri, E., Marhefka, G., and Vitale, M. 1996. Supravital exposure to propidium iodide identifies apoptotic cells in the absence of nucleosomal DNA fragmentation. Cytometry 23: 303.
2C. Third protocol: membrane permeability by Hoechst/PI
2C.1. Materials
-PBS, pH 7.0
-Hoechst (HO) 33342 1.5 ug/ml in PBS
-propidium iodide (PI) 5 ug/ml in PBS (always wear gloves when using
PI)
2C.2. Methodology
1. Incubate 1x106 cells with HO for 15 min at RT, then
stop the reaction on ice for 3 min.
2. Centrifuge the cells, discard the supernatant and resuspend the
pellet in 300-500 ul of PBS containing PI.
3. Analyze the sample by flow cytometry. Use a UV excitation (356
nm). Collect the blue (430 nm) fluorescence of HO and the red (630 nm)
fluorescence of PI.
3C. Commentary
3C.1. Background information
Apoptotic cells show a high HO staining and a low PI staining, since they initially tend to exclude PI. Necrotic cells are brightly stained with PI, while healthy cells are dimly stained by HO and not stained by PI.
3C.2. Time considerations
The whole procedure will take about 30 min.
3C.3. Key references
1. Belloc, F., Dumain, P., Boisseau, M. R., Jalloustre, C., Reiffers, J., Bernard, P., and Lacombe, F. 1994. A flow cytometric method using Hoechst 33342 and propidium iodide for simultaneous cell cycle analysis and apoptosis determination in unfixed cells. Cytometry 17: 59.
2. Ormerod, M. G., Sun, X. M., Snowden, R. T., Davies, R., Fearhead, H., and Cohen, G. M. 1993. Increased membrane permeability of apoptotic thymocytes: a flow cytometric study. Cytometry 14: 595.
2D. Fourth Protocol: DNA content
2D.1. Materials
-PBS pH 7.0
-Staining solution: a freshly prepared solution of (20 ug/ml propidium
iodide + 2 mg/ml DNAse-free RNAse in PBS) (always wear gloves when using
PI)
-70% ethanol
2D.2. Methodology
1. Fix the cells in suspension by adding 1 ml of cold (4°C)
70% ethanol to the resuspended pellet. Incubate 30 min on ice. Cells
can be stored in ethanol at -20°C up to 1 or 2 weeks.
2. Centrifuge the cells, and discard ethanol.
3. Resuspend the pellet in 300-500 ul of the staining solution, and
incubate for 15 min at RT.
4. Analyze by flow cytometry, at an excitation wavelength of 488
nm. Collect PI fluorescence at > 600nm.
3D. Commentary
3D.1. Background information
Fixation of cells with precipitating fixatives (such as ethanol or acetone) causes the leakage of the cleaved low MW DNA fragments that are produced during apoptosis. As a consequence, apoptotic cells can be identified as a hypodiploid peak, while healthy cells generate a typical cell cycle histogram. Necrotic cells are generally found among the healthy ones. The scatter methodology described before can help in the distinction between healthy and necrotic cells.
3D.2. Time considerations
The whole procedure will take about 1.5-2 hr.
3D.3. Key references
1. Nicoletti, I., Migliorati, G., Pagliacci, M. C., Grignani, F., and Riccardi, C. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Meth. 139: 271.
2. Zauli, G., Vitale, M., Re, M.C., Furlini, G., Falcieri, E., Gibellini, D., Visani, G., Davis, B.R., Capitani, S., La Placa, M. 1994. In vitro exposure to human immunodeficiency virus type 1 induces apoptotic cell death of the factor-dependent TF-1 hematopoietic cell line. Blood 83: 167.
APOPTOSIS vs. NECROSIS BY ELECTRON MICROSCOPY
1. Introduction
When analysed by light microscopy or fluorescence microscopy, (i.e. after Giemsa, Hoechst, DAPI, PI stainings and others) necrotic cells appear deeply different from apoptotic ones. The differences mainly concern cell shape and cell structural features. In numerous cell types frequently observable in culture and animal models, as well as human tissues, surface blebbing is considered a pattern specific of apoptosis. It is due to a deep cytoskeleton rearrangement, causing progressive changes in cell shape and organelle distribution. The final stage of this process is the cell splitting in numerous cellular portions, termed apoptotic bodies , whose most common final fate in vivo is to be engulfed by phagocytes. Blebs can be occasionally also described on the surface of cells undergoing necrosis, but, in this condition, they are followed by the rapid appearance of membrane discontinuties, causing water influx and strong ion distribution. No cell splitting appears in the course of necrosis, but a general cell hydration occurs, followed by cell swelling and disruption. Electron microscopy provides a detailed characterization of both phenomena, allowing the analysis of different cell compartments and, particularly, of the nucleus and its components.
Apoptotic and necrotic cells
can be studied and distinguished by TEM (transmission electron microscopy),
SEM (scanning electron microscopy) and FF (freeze-fracture), as well as
by means of some more specific and complex ultrastructural approaches (immuno-localization
of particular proteins, cytochemical localization of substrates, etc.)
(1).
1.TEM allows the analysis of sectioned specimens and provides a qualitative bidimensional image of the inner cell, after fixation, embedding, and staining.
2.SEM describes the cell surface and gives information about shape modifications and membrane specializations, with a relatively low detail resolution, but giving an overall information.
3.The FF technique utilizes frozen cells and the possibility to cleave them under high vacuum, exposing novel surfaces of the fractured cells, which are immediately platinum-carbon coated. These replicas, chemically purified from organic underlying tissue, are analyzed by the transmission electron microscope. New information on cell membrane and organelles can thus be obtained.
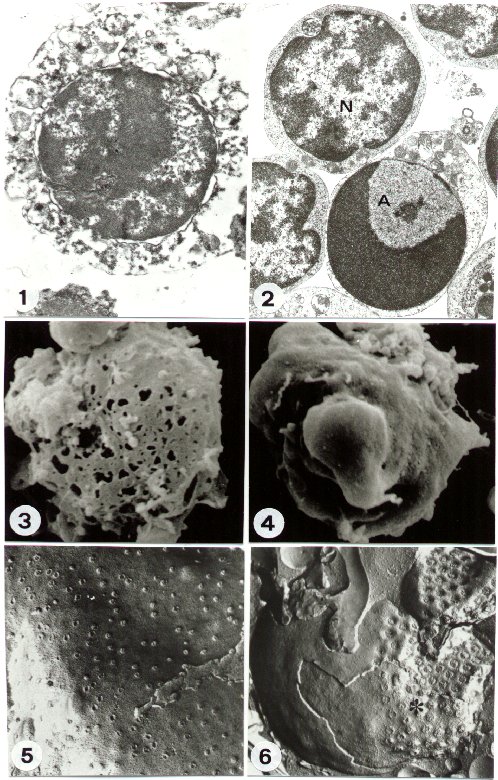
a) Necrosis: when a cell dies by necrosis the early changes can be identified on plasma membrane. It shows progressive discontinuities (Figure 1), which cause general cell hydration and swelling, as well as organelle disruption. This condition is clearly evidentiated by the early rearrangement of the freeze-fractured plasma membrane (2), as well as after SEM procedure of later necrotic stages (Figure 3).
The cytoplasm and plasma membrane are therefore the first target of the necrotic process, while the nucleus appears relatively well preserved, at least concerning its inner constituents (Figure 1), for some time.
b) Apoptosis: in the case of apoptotic death, the cell nucleus is early and specifically involved. TEM shows, indeed, a particular chromatin margination, followed by its compaction towards the nuclear perifery, to form one or several frequently cup-shaped masses. In the residual diffuse chromatin, remnants of deeply modified nucleoli can be still revealed (Figure 2). The nucleus appears therefore strongly rearranged, if compared to the normal one, which shows a perinuclear and a perinucleolar dense heterochromatin, clearly distinguishable from the diffuse interchromatin. (Figure 2). Surprisingly, plasma membrane and organelles are preserved for long, except for cytosol condensation and blebbing phenomenon, which characterize some apoptotic models (Figure 4). Subsequently, the nucleus generates numerous compact electron dense micronuclei, frequently released in the extracellular space. Cell splitting in a number of apoptotic bodies represents generally the final stage of apoptosis. The most common mechanism of apoptotic cell deletion in vivo is the engulfment by phagocytes. Differently, in vitro apoptotic cells undergo a late process of secondary necrosis.
FF reveals very peculiar
nuclear aspects in apoptotic cells. Cryoprotected frozen cells or tissues
can be indeed fractured in different possible ways. The nucleus can be
cleaved, thus showing chromatin arrangement and allowing the morpho-metrical
study of its fibres (3). More frequently, membrane leaflets are exposed,
and details of membrane architecture and specializations can be described.
In particular, the nuclear envelope of apoptotic cells displays
a characteristic clustering of nuclear pores (Figure 6), which are regularly
located - as TEM and FF comparison confirms - in close relationship of
the diffuse chromatin, being the dense chromatin masses pore-free. This
arrangement, never present in normal cells (Figure 5), has a specific functional
meaning and is typical of apoptotic cells (4).
2. Protocol
2.1. Materials
2.5% glutaraldehyde in phosphate buffer 0.1 M pH 7.3
1% osmium tetraoxide in phosphate buffer 0.1 M pH 7.3
0.2 M phosphate buffer solution (Sorensen)
0.15 M phosphate buffer solution (Sorensen)
alcohol-distilled water solutions 50%, 70%, 95%
alcohol 100%
propylene oxide
araldite resin: 50% component A, 50% component B, added with
1.5%
component C
1% toluidine blue in distilled water, added with 0.5% natrium carbonate
3% uranyl acetate in water: absolute alcohol, 1:1
lead citrate: 1.33 g lead nitrate + 1.76 g natrium citrate in 50
ml distilled water, added with 8 ml 1N NaOH
poly-L-lysine 1 mg/ml in distilled water
30% glycerol in 0.1 M phosphate buffer pH 7.3
freon 22
liquid nitrogen
commercial bleach
chloroform:methanol, 2:1
Reichert Ultracut FC4 Ultramicrotome
nickel or copper 100/200 mesh grids, coated with 4% formvar in chloroform
Balzers MED10 Critical Point Drying device
Balzers Gold Sputter device
Balzers BAF 400D Freeze-Fracture device
Philips CM10 Transmission Electron Microscope
Cambridge Stereoscan 200 Scanning Electron Microscope
2.2. Methodology
TEM
Observations with conventional transmission electron microscope are generally performed at 80 KV.
SEM
FF
3. Commentary
3.1. Background information
TEM procedures provide detailed information about necrotic and apoptotic cells, particularly in the case of very early and minimal changes, which are very difficult to detect with the other experimental approaches. Nevertheless, they are qualitative and do not generally provide quantitative information. Particularly, both necrosis and apoptosis, are generally focal phenomena and quantitation must be performed, before TEM, by light/ fluorescence microscopy or flow cytometry.
SEM can utilize very low magnification observations, so providing an overall picture of the specimen, but has some limits. Firstly SEM analysis is performed at low power resolution and, consequently, with a minor investigative potential. Secondly, it analyses the cell surface and no information about the inner cell are available. Finally, FF provides very detailed information. Pt-C thin replicas are indeed analyzed with a transmission electron microscope. This allows high magnifications and some possible morpho-functional correlations. In addition, the shadowing technique provides a 3-D image, which represents a complementary approach to conventional TEM. Unfortunatly, and in contrast with TEM sections, the fracture plane is casual and its direction (over or inside necrotic, apoptotic or normal cells) is unpredictable.
3.2. Critical Parameters
The most critical parameters for TEM, SEM and FF are the total number of cells necessary to process the samples and the percentage of necrotic or apoptotic cells present. This can be previously checked, in similar experimental conditions, by flow cytometry.
3.3. Troubleshooting
A good and immediate fixation, to which a particular care must be devoted, is crucial.
3.4. Time considerations
Complete TEM procedure takes 4-5 days, SEM takes 2 days and FF takes 1 day. The time spent for the electron microscope observation is variable and strogly dependent on the operator s experience.
3.5. Key References
FIGURE 1
TEM of a necrotic cell: the disruption of plasma membrane and organelles
is observable. A relative preservation of nuclear morphology appears.
(original magnification: x 10,000)
FIGURE 2
TEM of an apoptotic (A) and a normal (N) cell. The characteristic chromatin
rearrangement appears in A, strongly different from its normal organization
(N). The good preservation of membrane and organelles is also evident.
(original magnification: x 8,000)
FIGURE 3
SEM of a necrotic cell. Numerous lesions appear on the cell surface.
(original magnification: x 5,000)
FIGURE 4
SEM of an apoptotic cell. Surface blebbing is evident. (original
magnification: x 5,000)
FIGURE 5
FF of normal cell, nuclear envelope. The regular distribution of nuclear
pores is visible. (original magnification: x 30,000)
FIGURE 6
FF of apoptotic cell. The nuclear envelope shows a characteristic clustering
(asterisc) of nuclear pores. (original magnification:
x 35,000)