
Ruggero De Maria*, Ann Zeuner*, Angela Santoni# and Roberto Testi*.
*Department of Experimental Medicine and Biochemical Sciences, "Tor
Vergata"
University, #Department of Experimental Medicine and Pathology,
"La Sapienza" University, Rome, Italy.
The role of Ca2+ signaling:
from activation to apoptosis
Ca2+ is the most common signal transduction element in cells ranging from bacteria to neurons and it is involved in such diverse cellular processes as fertilization, muscle contraction, cell growth, and apoptosis. Intracellular calcium concentration must be kept at very low levels (± 100 nM, 20,000 fold lower than the 2 mM found extracellularly) since this ion precipitates phosphates, the energy currency of cells; cytosolic Ca2+ concentrations are therefore susceptible of rapid and localized increases, which are obtained simply letting the calcium ions enter by gradient density across the plasma membrane or from the ER through specialized channels (1).
In nonexcitable cells, such as blood cells, there are two mechanisms to introduce small bursts of Ca2+ into the cytosol for signal transduction, both of which lead to the formation of InsP3 from the hydrolysis of an inositol lipid precursor stored in the plasma membrane. One pathway is initiated by a family of G protein-linked receptors (to which belong adrenergic, muscarinic, serotonin receptors and many others), that activate PLCb1 via a GTP binding protein and cause the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce InsP3 and diacylglycerol (DAG). The other pathway responsible for stimulating the release of InsP3 begins with tyrosine kinase receptors (as PDGFR or EGFR), which undergo ligand mediated dimerization and phosphorylate each other on specific tyrosine residues, providing docking sites for phospholipase Cg1 (PLCg): in unstimulated cells, PLCg is largely cytosolic, but translocates to the membrane as its SH2 domain binds to the activated receptor. This association has two important consequences for the activation of PLCg: it is phosphorylated by the receptor on specific tyrosine residues, while its membrane translocation brings it in contact with its substrate PIP2 (2). A similar process is responsible for stimulating PLCg following crosslinking of IgM receptors in B lymphocytes, or for activating the T-cell antigen receptor (TCR)-CD3 complex in T cells; although these lymphocyte receptors lack intrinsic tyrosine kinase activity, they are able to recruit members of the src family such as fyn or lck which in turn interact with PLCg (18).
The activation of PLCg results in the hydrolysis of PIP2 yielding to the formation of the second messengers InsP3 and DAG, which are responsible for a sustained increase in cytoplasmic free Ca2+ and activation of protein kinase C, respectively. Although the second messengers that are produced are the same in both pathways, in general tyrosine kinase-activated PLCgs increase Ca2+ more slowly and for longer duration than do G protein-mediated PLCbs.
Once PLC splits PIP2 into InsP3 and DAG, a complex set of enzymes mediates the generation of multiple inositol phosphates, some of which can influence calcium influx (4), but undoubtedly InsP3 is the dominant second messenger molecule for the release of intracellular Ca2+ (2). InsP3 receptor is a homotetramer surrounding a relatively nonselective cationic pore (5), and it is found on the surface of the Ca2+-storing organelles and in some cases (for example in lymphocytes) on the plasma membrane (6, 7). Regulation of the InsP3 receptor is complex, in that it binds multiple InsP3 molecules, is desensitized by InsP3 itself (8), is phosphorylated by protein kinase A, and has a biphasic sensitivity to cytoplasmic Ca2+ levels (9, 10).
The rise in [Ca2+]i evoked by binding of InsP3 to its receptor is typically biphasic: the initial rise results from InsP3 -mediated opening of channels on the surface of intracellular calcium stores; this release of Ca2+ ions is transient in nature and is usually followed by a more sustained elevation of the intracellular [Ca2+]i due to Ca2+ entry across the plasma membrane: it appears that Ca2+ entry across the plasma membrane is somehow coupled to the depletion of intracellular calcium stores by InsP3 ; this process is a field of intense investigation and is called "capacitative calcium entry" (11). Many putative ion channels eventually responsible for the second phase of calcium signaling have been identified, but the most well-established pathway seems to be ICRAC (Ca2+ release-activated current), which has an extremely low conductance and is activated by several experimental procedures that result in depletion of stores (12). A number of second messengers have been proposed to initiate ICRAC, including small G proteins, pertussis toxin-sensitive G proteins, cGMP, various lipids, tyrosine kinases, and InsP3 , but to date no conclusive findings have been reported. The most interesting and controversial candidate second messenger for capacitative entry is Ca2+ influx factor (CIF), that has been partially characterized as a phosphorylated pH-stable anion (13); presumably CIF is released or generated from the ER or adjacent regions in response to InsP3 -mediated Ca2+ release from stores, but its exact nature is still to be established (14). Key issues to be settled in this field are the concrete identification of either a second messenger or a direct signaling that mediates capacitative Ca2+ entry through the extracellular space, and whether there is more than one Ca2+ entry pathway mediated by store depletion.
Cellular responses to increased [Ca2+]i have been extensively studied in T lymphocytes, where the calcium/calcineurin pathway is linked to the regulation of IL-2 gene expression: calmodulin-complexed cytoplasmic Ca2+ activates calcineurin, a serine phosphatase implicated in the activation of transcription factors that bind to the antigen receptor response elements within the IL-2 enhancer region upon T cell activation. The calcium signaling pathway in T lymphocytes is blocked by the immunosuppressive drugs cyclosporine A (CsA) and FK506, whose clinical efficacy has revolutionized the field of organ transplantation: both CsA and FK506 bind to cytoplasmic proteins, known as immunophilins, and the drug-immunophilin complex interacts at high affinity with calcineurin interfering with its enzymatic function and so inhibiting the transcription of several lymphokyne genes (15).
IL-2 gene activation is definitely not the only example of Ca2+ influence on gene transcription: intranuclear Ca2+ increases initiate gene expression in a way that depends both on the route of calcium entry and on cellular Ca2+ levels (16, 17). Activation of definite transcription factors by elevated [Ca2+]i also affects cell cycle progression in many experimental systems: for example, calcineurin, the Ca2+-activated Ser/Thr phosphatase, is required for G0 to G1 transitions in yeast and mammalian cells, and Ca2+ is necessary and sufficient for resumption of meiosis in marine eggs (18). However, understanding of the specific mechanisms for these effects is still at an early stage.
Variations in cytosolic Ca2+ content can also activate degradative processes in programmed cell death, or apoptosis. Increases in [Ca2+]i can signal for initiation of apoptosis - possibly by direct stimulation of calcium-dependent endonucleases that are responsible for DNA degradation in some cells such as thymocytes, while in other experimental systems calcium chelators promote cell death. The mechanisms through which Ca2+ influences apoptotic processes are not understood, and are not an easy field of investigation in that the results are often completely different from one cell type to one other. Nevertheless, increased amounts of type 3 InsP3 receptors have been detected on the plasma membrane of B and T lymphocytes undergoing apoptosis. This translocation alters the [Ca2+]i and seems causally linked to the induction of cell death (19). Moreover, there are some evidences that calcineurin, calmodulin, and the calcium-dependent cysteine protease calpain may play important roles in mediating the upstream events in programmed cell death (20-22). Finally, AGL-2, an apoptosis-related gene that has been recently identified codes for a Ca2+-binding protein and is a central component of both T cell receptor- and glucocorticoid cell death (23).
Understanding the role of calcium ions in transcriptional control, cell division and cell death is one of the most important and complex areas of research in immunology, and requires appropriate tools of investigation.
New tools for studying receptor-mediated Ca2+ fluxes
A new and widely used method to measure kinetic variations of [Ca2+]i in specific cell subpopulations, identified by surface immunofluorescence staining with specific antibodies (Abs), has been recently described. The method, developed by Tsien and coll., bases on the use of the Ca2+ indicator Fluo-3, a fluorescein derivative which undergoes a dramatic increase in fluorescence intensity upon Ca2+ binding (24). Fluo-3 has emission/excitation spectra similar to those of fluorescein, and displays minimal wavelength shifts following Ca2+ binding, being particularly suitable for the use of cytofluorimeters equipped with argon-ion laser and fluorescein optical filters (25).
Due to the nature of their natural ligands, the study of Ca2+ fluxes in hematopoietic cells, following engagement of surface receptors, often requires intense crosslinking. With the exception of a few major receptors, including CD3, CD2, CD16 and the BCR, most surface receptors are not able to induce [Ca2+]i increase when triggered by their specific mAbs in combination with a secondary Ab.
We describe here a simple cytofluorimetric method to measure [Ca2+]i levels following extensive crosslinking of surface receptors. The procedure takes advantage from the use of sulfate polystyrane latex beads bound to mAbs, or to the natural ligands of the receptors to be studied, and has been successfully employed for studying the early signals generated by CD69, CD44 and b1 integrins (26-28).
A simple method for extensive receptor crosslinking and [Ca2+]i measurement
Preparation of Ab coupled beads. Wash 200 ml of polystyrene latex beads (2.3 mm diameter; IDC, Portland, OR) twice in carbonate buffer (30 mM Na2CO3 and 70 mM NaHCO3, pH 9.5), and incubate the beads with 50-200 mg of purified mAbs or proteins, in 300 ml of carbonate buffer, for 30-60 min at room temperature and under continuous agitation. Wash the beads three times with PBS and resuspend them in 2 ml of PBS without calcium and magnesium ions.
Ab-bound beads can be sterilized in 0.01% formaldehyde for 30 min and, after extensive washing in sterile PBS, stored for up to 1 year at 4° C. Natural ligands or control proteins are preferably used shortly after conjugation to the beads. The amount of mAb or protein bound to the beads can be easily monitored by fluorescent Ab staining and flow cytometry analysis. To this purpose, both control and stimulatory beads are first incubated with 5% BSA for 20 min at RT, and then stained with anti-coated protein (Ab or natural ligand) specific fluorescent Ab.
Cell labeling. Cells are first washed in RPMI containing 1% FCS and incubated (up to 10 x 106/ml) with 1-4 mM Fluo-3 acetoxymethyl ester and 1 mg/ml Pluronic F-127 (Molecular Probes, Inc., Europe BV, ND), dissolved in dimethylsulfoxide, for 30-45 min at 37° C in the dark, with gentle shaking every ten min. After three washings with warm medium, which allow cellular esterases to act on Fluo-3, cells are ready for flow cytometry. If a particular subset of the whole cell population needs to be analyzed, cells are stained with phycoerythrin- or PerCP- (Becton Dickinson, San Jose, CA) or Cy-Chrome- (Pharmingen, San Diego, CA) conjugated mAb, specific for the cell subsets to be studied, performing a two- or three-color analysis. After labeling, cells can be stored on ice for one or two hours before the [Ca2+]i measurement is performed.
Ca2+ determination. After Fluo-3 loading, cells are kept at 37° C for 10 min, and run on the flow cytometer for the determination of basal fluorescence in unstimulated cells. Cells are then mixed with 50 ml of beads, spin for 8 sec at 10,000 g, and immediately acquired on the flow cytometer. The acquisition intervals on the green (525) channel should be ~ 10 sec, and the number of events to be acquired should be ~ 3,000. The kinetics may vary considerably from experiment to experiment. To convert fluorescence values into absolute [Ca2+]i, a calibration procedure is required on each experiment (24). [Ca2+]i is therefore calculated using the equation:
[F - Fmin]
[Ca2+]i = Kd x -------------------
[Fmax - F]
where the Kd of Fluo-3 is 400 nM, F is the sample mean fluorescence, and Fmax is obtained by exposing the cells in 1 mM Ca2+ ([Ca2+]e can be easily adjusted by adding calcium chloride to a Ca2+- and Mg2+-free medium) to 5 mg/ml ionomycin. To obtain Fmin, ionomycin-treated cells are exposed to 2 mM manganese chloride. Mn2+ displaces Ca2+ from Fluo-3, forming a complex eight-fold more fluorescent than the metal-free dye, but five times lower than the Ca2+ / Fluo-3 complex. Therefore, Fmin can be calculated as follows:
Fmin = 1.25 x FMnCl2 - 0.25 x Fmax
The acquisition mode can be either linear or
logarithmic. No conversion from logarithmic to linear values is required
when mean fluorescence intensity is converted to absolute [Ca2+]i
content.
Studying CD69-generated Ca2+ influx in hematopoietic cells: a common signal for multiple functions
CD69 is a type II membrane protein member of the natural killer cell gene complex family of cell surface receptors. This receptor is widely distributed on all hematopoietic cell types, as monocytes and platelets constitutively express CD69, whereas lymphocytes and granulocytes express CD69 shortly following cellular activation (29-32). Molecular crosslinking of CD69 receptors induces rapid and sustained extracellular Ca2+ influx, responsible for initiating disparate cellular functions. In fact, such a broad distribution associates to diverse functions triggered by CD69 in different cell types, including lymphocyte proliferation and cytotoxicity, platelet aggregation, macrophage cytotoxicity and apoptosis, granulocyte activation and production of inflammation mediators (33).
In most cell types CD69 requires extensive crosslinking to effectively transduce an effective stimulatory signal. Accordingly, Ca2+ influx is usually not generated by soluble mAbs. However, stimulation by anti-CD69-coupled beads resulted in elevated and extended [Ca2+]i increase, due to a massive Ca2+entry across the plasma membrane (Fig. 1).
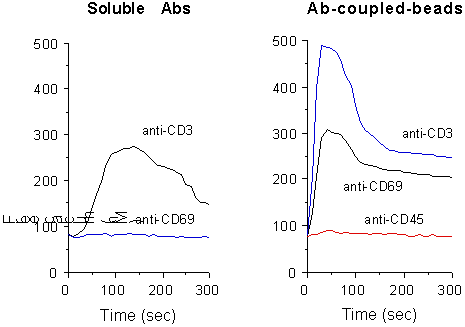
Fig. 1. Measurement of [Ca2+]i
content in PMA-stimulated T cells, following CD69 and CD3 crosslinking
by soluble GAM (left) or mAb-conjugated beads (right). Y axes show [Ca2+]i
contents (nM).
A complex balance of kinases and phosphatases regulates receptor-generated signals, modulating the level of substrate phosphorylation (3). CD69 is constitutively phosporylated on serine residues (33). To determine whether the levels of serine phosphorylation are able to influence the capacity of CD69 triggering to act on plasma membrane ion channels, we treated activated CD69 positive T cells either with okadaic acid, an inhibitor of serine phosphatases , or with staurosporin, a potent serine kinases inhibitor. As shown in Fig. 2, CD69 phosphorylation inversely correlates with its ability to induce [Ca2+]i increase upon receptor crosslinking, suggesting that serine phoshporylation controls CD69 responsiveness by modulating its ability to generate activation signals.
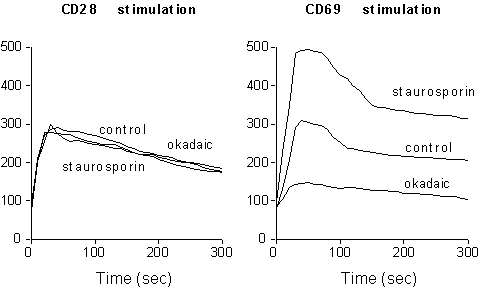
Fig. 2. Effect of serine phosphorylation on
CD69-induced Ca2+-signaling. PMA treated T cells were
incubated for two hours with either 50 nM okadaic acid or 20 nM staurosporin,
loaded with Fluo-3, and stimulated with anti-CD28 or anti-CD69-coupled
beads. Y axes show [Ca2+]i contents
(nM).
Thus, Ab-coupled beads constitute an essential
tool for studying the regulation of signals induced by CD69 stimulation,
as well as by a number of other receptors requiring extensive crosslinking
to reproduce in vitro the effect of their natural ligands.
REFERENCES
1. Clapham, D. E. 1995. Calcium signaling. Cell 80:259-268.
2. Berridge, M. J. 1993. Inositol triphosphate and calcium signalling. Nature 361:315-325.
3. Weiss, A., and D. R. Littman. 1994. Signal transduction by lymphocyte antigen receptors. Cell 76:263-274.
4. Berridge, M. J., and R. F. Irvine. 1989. Inositol phosphates and cell signalling. Nature 341:197-205.
5. Mikoshiba, K. 1993. Inositol 1,4,5-receptor. Trends Pharmacol. Sci. 14:86-89.
6. Fujimoto, T., S. Nakade, A. Miyawaki, K. Mikoshiba, and K. Ogawa. 1992. Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J. Cell. Biol. 119:1507-1513.
7. Khan, A. A., J. P. Steiner, M. G. Klein, M. F. Schneider, and S. H. Snyder. 1992. IP3 receptor: localization to plasma membrane of T cells and cocapping with the T cell receptor. Science 257:815-818.
8. Hajnoczky, G., and A. P. Thomas. 1994. The inositol triphosphate calcium channel is inactivated by inositol triphosphate. Nature 370:474-477.
9. Finch, E. A., and S. M. Goldin. 1994. Calcium and inositol 1,4,5-triphosphate-induced Ca2+ release. Science 265:813-815.
10. Bezprozvanny, I., J. Watras, and B. E. Ehrlich. 1991. Bell-shaped calcium-response curves of Ins(1,4,5)P3 and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751-754.
11. Putney, J. W. 1993. The signal for capacitative calcium entry. Cell 75:199-201.
12. Fasolato, C., B. Innocenti, and T. Pozzan. 1994. Receptor-activated calcium influx: how many mechanisms for how many channels? Trends Pharmacol. Sci. 15:77-83.
13. Randriamampita, C., and R. Y. Tsien. 1993. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature 364:809-814.
14. Clapham, D. E. 1994. A mysterious new influx factor? Nature 364:763-764.
15. Liu, J., J. D. J. Farmer, W. S. Lane, J. Friedman, I. Weissman, and S. L. Schreiber. 1991. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807-815.
16. Negulescu, P. A., N. Shastri, and M. D. Cahalan. 1994. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc. Natl. Acad. Sci. USA 91:2873-2877.
17. Bading, H., D. D. Ginty, and M. E. Greenberg. 1993. Regulation of gene expression in hippocampal neurons by dinstinct calcium signalling pathways. Science 260:181-186.
18. Means, A. R. 1994. Calcium, calmodulin and cell cycle regulation. FEBS Lett. 347:1-4.
19. Khan, A. A., M. J. Soloski, A. H. Sharp, G. Schilling, D. M. Sabatini, S. H. Li, C. A. Ross, and S. H. Snyder. 1996. Lymphocyte apoptosis: mediation by increased type 3 inositol 1,4,5-trisphosphate receptor. Science 273:503-507.
20. Shibasaki, F., and F. McKeon. 1995. Calcineurin functions in Ca(2+)-activated cell death in mammalian cells. J. Cell. Biol. 131:735-743.
21. Sarin, A., D. H. Adams, and P. A. Henkart. 1993. Protease inhibitors selectively block T cell receptor-triggered programmed cell death in a murine T cell hybridoma and activated peripheral T cells. J. Exp. Med. 178:1693-1700.
22. Squier, M. K., A. C. Miller, A. M. Malkinson, and J. J. Cohen. 1994. Calpain activation in apoptosis. J. Cell. Physiol. 159:229-237.
23. Vito, P., E. Lacana', and L. D'Adamio. 1996. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science 271:521-525.9.
24. Kao, J.P., A. T. Harootunian, and R. Y. Tsien. 1989. Photochemically generated cytosolic calcium pulses and their detection by fluo-3. J. Biol. Chem. 264, 8179-8184.
25. Vandenberghe, P.A. and J. L. Ceuppens. 1990. Flow cytometric measurement of cytoplasmic free calcium in human peripheral blood T lymphocytes with Fluo-3, a new fluorescent calcium indicator. J. Immunol. Methods 127, 197-205.
26. Testi, R., J. H. Phillips, and L. L. Lanier. 1989. T cell activation via Leu23 (CD69). J. Immunol. 143:1123-1128.
27. Galandrini, R., R. De Maria, M. Piccoli, L. Frati, and A. Santoni. 1994. CD44 triggering enhances human NK cell cytotoxic functions. J. Immunol. 153, 4399-4407.
28. Palmieri, G., A. Serra, R. De Maria, A. Gismondi, M. Milella, M. Piccoli, L. Frati, and A. Santoni. 1995. Crosslinking of a4b1 and a5b1 fibronectin receptors enhances NK cytotoxic activity. J. Immunol. 155, 5314-5322.
29. De Maria, R., M. G. Cifone, R. Trotta, M. R. Rippo, C. Festuccia, A. Santoni, and R. Testi. 1994. Triggering of human monocyte activation through CD69, a member of the natural killer cell gene family of signal transducing receptors. J. Exp. Med. 180:1999-2004.
30. Testi, R., F. Pulcinelli, L. Frati, P. P. Gazzaniga, and A. Santoni. 1990. CD69 is expressed on platelets and mediates platelet activation and aggregation. J. Exp. Med. 172:701-707.
31. De Maria, R., S. Fais, M. Silvestri, L. Frati, F. Pallone, A. Santoni, and R. Testi. 1993. Continuous in vivo activation and transient hyporesponsiveness to TCR/CD3 triggering of human gut lamina propria T lymphocytes. Eur. J. Immunol. 23:3104-3108.
32. Gavioli, R., A. Risso, D. Smilovich, I. Baldissarro, M. C. Capra, A. Bargellesi, and E. Cosulich. 1992. CD69 molecule in human neutrophils: its expression and role in signal-transducing mechanisms. Cell. Immunol. 142:186-196.
33. Testi, R., D. D'Ambrosio, R. De Maria,
and A. Santoni. 1994. The CD69 receptor: a multipurpose cell surface
trigger for hematopoietic cells. Immunol. Today 15:479-483.
